
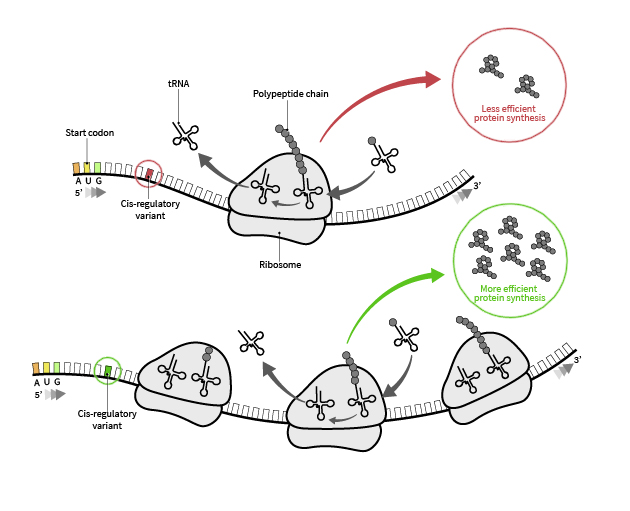
Advances in sequencing have greatly accelerated our ability to catalog human genetic diversity, yet the functional consequences of these genomic differences are still poorly understood. Genetic variants typically exert phenotypic effects through impacts on protein function and abundance. By integrating RNA-seq data with transcription factor binding and chromatin profiles, significant progress has been made to our understanding of the genetic basis of RNA expression. As we and others have shown, however, protein levels are often poorly explained by RNA expression alone, suggesting additional critical regulatory steps. Although translation efficiency, i.e. the number of proteins synthesized per transcript, contributes significantly to determining protein levels, little is currently known about the genetic determinants of translation efficiency in humans or their relevance to disease.
For my postdoctoral work at Stanford, I have focused on the human genetics of translation regulation, generating RNA-Seq and ribosome profiling data from a diverse set of individuals and integrating these measurements with quantitative mass spectrometry-based proteomics. This study revealed that translation efficiency indeed varies among individuals and that the genetic factors underlying this translational variability can be mapped. In several cases, we pinpointed the mechanisms controlling the observed variation in translation efficiency, with genetic variability in sequences near the translation initiation sites being one such example.
However, lack of sensitive methods limits our ability to characterize a potentially large class of regulatory variants that modulate translation. A major objective in our lab is to develop a suite of experimental and computational tools that will identify cis-acting genetic regulators of translation. To address these limitations, our laboratory will undertake two approaches. First, in an interdisciplinary collaboration, we designed and built a custom microfluidic chip that enables simultaneous purification and selection of RNA fragments protected by ribosomes (17-35nt) using isotachophoresis. We coupled this innovation with the latest advances in small RNA sequencing library preparation and optimization of RNase digestion conditions. Our preliminary data established the feasibility of generating high quality ribosome profiling data from as few as 1000 cells. Our laboratory will further develop this approach to yield a groundbreaking technology that will reveal the translation landscape of a wide range of biologically important samples.
Second, to determine cis-acting genetic regulators of translation, we will use allele-specific analyses. By comparing relative translation of two alleles from the same cell population, we will ensure that the genetic background, environmental factors and sample preparation are identical. This strategy has proven extremely powerful in identifying genetic variants that control RNA expression. Specifically, we will develop and distribute a suite of bioinformatics tools that will robustly generate allele-specific translation candidates using RNA sequencing and ribosome profiling data. We will complement these approaches with an experimental assay for measuring allele-specific translation.